

Co-manufacturing of Veterinary and Human Vaccines in the Same Facility
Co-manufacturing of Veterinary and Human Vaccines
in the Same Facility
A Risk-Stratified Global GMP Analysis
Differentiating by Vaccine Type, Biological Agent Class, and Manufacturing Scope
Regulatory Affairs & Quality Systems Review | June 2026
ABSTRACT
Whether veterinary and human vaccines may be manufactured in the same facility is not answerable by a single binary response under any major global GMP framework. The permissibility — and the stringency of controls required — varies substantially depending on three intersecting risk dimensions: (1) whether the vaccine contains live attenuated organisms or killed/inactivated agents; (2) whether the organisms are viral or bacterial in nature, and their associated pathogenicity, zoonotic potential, and resistance to decontamination; and (3) whether the manufacturing scope encompasses full upstream biological production, downstream purification, or only aseptic fill/finish operations on an already-inactivated drug substance. This article applies a risk-stratified framework grounded in the EU GMP EudraLex Volume 4 (Annexes 2 and 5), WHO Technical Report Series 978/986, PIC/S PE 009-17 and PS/INF 85/2021, WOAH Terrestrial Manual Chapters 2.3.3 and 1.1.8, ICH Q9(R1), and the US FDA/USDA and UK MHRA frameworks to evaluate six representative co-manufacturing scenarios, determine their regulatory permissibility, and define the minimum controls required in each case.
1. Introduction
The previous iteration of this analysis established that no global GMP framework categorically prohibits the co-location of veterinary and human vaccine manufacturing, but that all impose demanding conditions. However, a critical limitation of that analysis was its treatment of ‘vaccine manufacturing’ as a monolithic activity. In practice, the regulatory risk profile of a shared facility is inseparable from three fundamental variables that regulators explicitly evaluate, yet the prior article did not systematically differentiate:
- The biological status of the vaccine antigen: whether the organism is live-attenuated (still viable and potentially transmissible) or killed/inactivated (rendered non-replicating by validated physical or chemical means);[1][2]
- The taxonomic class of the agent: whether it is a virus or bacterium, and in either case its pathogenicity, zoonotic potential, host range, resistance to disinfection, and classification under national biosafety frameworks;[3][4]
- The manufacturing scope: whether both upstream (fermentation/cell culture, virus propagation) and downstream (purification, inactivation, formulation) activities are performed, or whether the facility operates solely as a fill/finish site receiving an already-inactivated, purified drug substance.[5][6]
These three dimensions interact multiplicatively to determine the overall risk level of a co-manufacturing arrangement. A fill/finish facility handling two killed viral drug substances — one for humans, one for animals — presents a fundamentally different risk profile than an upstream biological production facility growing live attenuated viral strains of zoonotic potential for both species. Regulatory GMP frameworks, when read carefully, implicitly or explicitly recognize this gradation. This article makes that gradation explicit.
2. The Three-Dimensional Risk Framework
2.1 Dimension 1: Biological Status — Live Attenuated vs. Killed/Inactivated
The distinction between live attenuated and killed vaccines is the single most consequential variable in determining the GMP risk of shared manufacturing.
Live attenuated vaccines contain organisms — viral or bacterial — that have been weakened by serial passage or genetic modification to reduce pathogenicity while preserving immunogenicity. They remain biologically viable: capable of replication, mutation, and — in the case of organisms with zoonotic potential — potentially crossing species barriers. The quality and safety requirements for live attenuated vaccines are higher than for killed and subunit vaccines. [1] They require Biosafety Level (BSL)-2 or BSL-3 production environments throughout the upstream phase, negative-pressure containment to prevent environmental release, and no concurrent handling of different strains in the same area. [3]
Killed/inactivated vaccines undergo a validated chemical (e.g. formaldehyde, beta-propiolactone) or physical (heat, UV) inactivation step that renders the organism non-replicating. Once inactivation has been validated and confirmed — typically via specific inactivation kinetics studies — the biological hazard of the drug substance is fundamentally transformed. [7] Post-inactivation materials are generally handled in clean areas rather than containment zones, and the risk of transmission of viable organisms is eliminated, provided inactivation validation is robust. The inactivation step is thus a critical process control that changes the risk tier of the product — and by extension, the permissibility of shared facilities — at the point in the manufacturing process at which it occurs.
⚠ REGULATORY PRINCIPLE: EU GMP Annex 5 and WOAH Chapter 2.3.3 both explicitly differentiate containment requirements by biological agent status: live agents must be in contained areas; inactivated agents may be handled in clean areas. This distinction directly maps to shared facility permissibility.
2.2 Dimension 2: Viral vs. Bacterial Vaccines — Agent-Specific Risk Factors
The biological classification of the vaccine agent introduces further risk differentiation.
2.2.1 Viral Vaccines
Viral vaccine production typically involves the infection of cell culture systems (Vero cells, MRC-5, CHO, insect cells) with the viral agent. Viruses present specific contamination risks including:
- Adventitious virus contamination: the introduction of unintended extraneous viruses through raw materials, cell banks, or the environment, which may be difficult to detect and may not be eliminated by the inactivation step designed for the target agent.[7]
- Viral vector cross-contamination: recombinant viral vaccine platforms (adenoviral vectors, poxvirus vectors) may recombine with helper viruses or other genetic elements if incompletely segregated.[8]
- Zoonotic risk of animal-derived viruses: veterinary vaccines may employ animal viruses with unknown or emerging zoonotic potential. The BSL classification of the organism drives facility design requirements.[4][9]
- Reversion to virulence: for live attenuated viral vaccines, particularly oral poliovirus vaccine and certain flavivirus vaccines, the risk of reversion to wild-type through back-mutation is a specific manufacturing hazard requiring negative-pressure containment and rigorous environmental monitoring.[3][10]
Live viral vaccines require facilities meeting BSL-2 as a minimum; vaccines derived from more pathogenic organisms (e.g. influenza H5N1, rabies, polio, SARS-CoV-2) require BSL-2 enhanced or BSL-3 conditions. Inactivated viral vaccines require BSL-2 upstream (pre-inactivation), dropping to BSL-1 post-validated inactivation. [3,9]
2.2.2 Bacterial Vaccines
Bacterial vaccine production — whether live attenuated (BCG, Ty21a, CVD 103-HgR) or killed (pertussis, cholera whole-cell, anthrax) — presents distinct challenges:
- Spore-forming bacteria (e.g. Clostridium, Bacillus anthracis): present an extreme decontamination challenge. EU GMP Annex 5 and WOAH Chapter 2.3.3 explicitly require that areas handling spore-forming bacteria be separated and dedicated until the agents are inactivated. Shared manufacture is effectively prohibited at the upstream level.[11][12]
- Non-spore-forming bacteria (e.g. BCG, Salmonella Typhi Ty21a, Vibrio cholerae): while less environmentally persistent than spore-formers, live attenuated bacterial vaccines still require careful segregation. The BCG vaccine is specifically cited in PIC/S GMP Annex 2 as requiring dedicated facilities.[2]
- Bacterial endotoxins: the presence of lipopolysaccharide (LPS) from Gram-negative bacteria presents a product-quality cross-contamination risk even after inactivation, requiring validated depyrogenation procedures and stringent endotoxin testing.[13]
- Antimicrobial resistance: some bacterial vaccine strains carry genetic resistance markers for selection purposes. Cross-contamination of human products with veterinary bacterial strains carrying such markers carries regulatory and safety implications.[2]
2.3 Dimension 3: Manufacturing Scope — Upstream, Downstream, and Fill/Finish
The third dimension — what manufacturing activities are actually performed at the shared site — is operationally decisive. The risk gradient across the manufacturing value chain is substantial and directly maps to the regulatory requirements that apply.
2.3.1 Upstream Manufacturing (Fermentation / Cell Culture / Virus Propagation)
Upstream manufacturing is where the biological agent is grown, propagated, and amplified. It is the phase of highest biological risk: organisms are live and at high concentrations; aerosol generation from bioreactors is a meaningful hazard; and environmental contamination, if it occurs, involves viable and potentially transmissible agents. [5,6]
All major GMP frameworks impose their strictest containment requirements at this stage: BSL-appropriate facility design (including negative pressure for live attenuated or highly pathogenic agents), dedicated bioreactor suites, HEPA-filtered exhaust, validated effluent inactivation, and strict personnel decontamination procedures. [11,12]
Co-manufacturing of human and veterinary vaccine upstream activities — particularly involving live attenuated organisms — is subject to the highest regulatory scrutiny and is, in many scenarios, incompatible with the use of shared areas.
2.3.2 Downstream Manufacturing (Purification, Inactivation, Formulation)
Downstream processing begins at the point of harvest of the crude biological material and includes inactivation, clarification, purification, concentration, and formulation. The critical regulatory inflection point within this phase is the validated inactivation step.
Prior to confirmed inactivation, the material is handled as if it were at the same biological risk level as the upstream product. Once inactivation has been validated — typically requiring demonstrated kinetics showing a specified log reduction of infectivity — the product’s BSL classification effectively reverts, and requirements for containment are correspondingly reduced. [7,8]
Post-inactivation downstream operations (e.g. chromatography, ultrafiltration, formulation) may, in principle, be carried out in shared areas, provided that: (1) the inactivation step is rigorously validated; (2) campaign separation with validated cleaning is maintained; (3) a QRM assessment documents the residual risks; and (4) the products have compatible Health-Based Exposure Limits (HBELs). [13]
2.3.3 Fill/Finish Operations Only
Fill/finish refers to the final aseptic filling of the drug product (already formulated drug substance) into its primary container — vials, syringes, or ampoules — and subsequent closure, labelling, and packaging. Where a facility operates exclusively as a fill/finish site — receiving a pre-formulated, inactivated, purified drug substance from a separate upstream/downstream manufacturing site — the biological hazard profile is fundamentally different from an integrated production facility.
At the fill/finish stage, assuming the drug substance has been appropriately inactivated and purified, no live pathogenic organism is present. The contamination risks are primarily:
- Microbiological contamination of the sterile product during aseptic filling — addressed by Annex 1 requirements (Contamination Control Strategy, Grade A/B environments, environmental monitoring);[14]
- Mix-up of products with different species or antigen specificities;[6]
- Cross-contamination of residual protein antigens between products — evaluated via HBEL-based cleaning validation.[13]
Fill/finish co-manufacturing of human and veterinary vaccines — particularly killed/inactivated products — is the scenario most likely to be permissible under global GMP frameworks, provided robust aseptic controls and campaign manufacturing with validated cleaning are in place. The absence of live biological agent at this stage removes the containment imperatives that drive separation requirements at earlier manufacturing stages.
✔ KEY PRINCIPLE: The inactivation step is the pivotal risk boundary in the manufacturing process. Before validated inactivation: live-agent containment rules apply in full. After validated inactivation: aseptic/clean-room GMP rules govern. This principle, embedded in EU GMP Annex 5, WOAH Ch. 2.3.3, and WHO TRS 978, is the regulatory foundation for a graduated, risk-based approach to shared facilities.
3. Application of Global GMP Frameworks to Risk-Stratified Scenarios
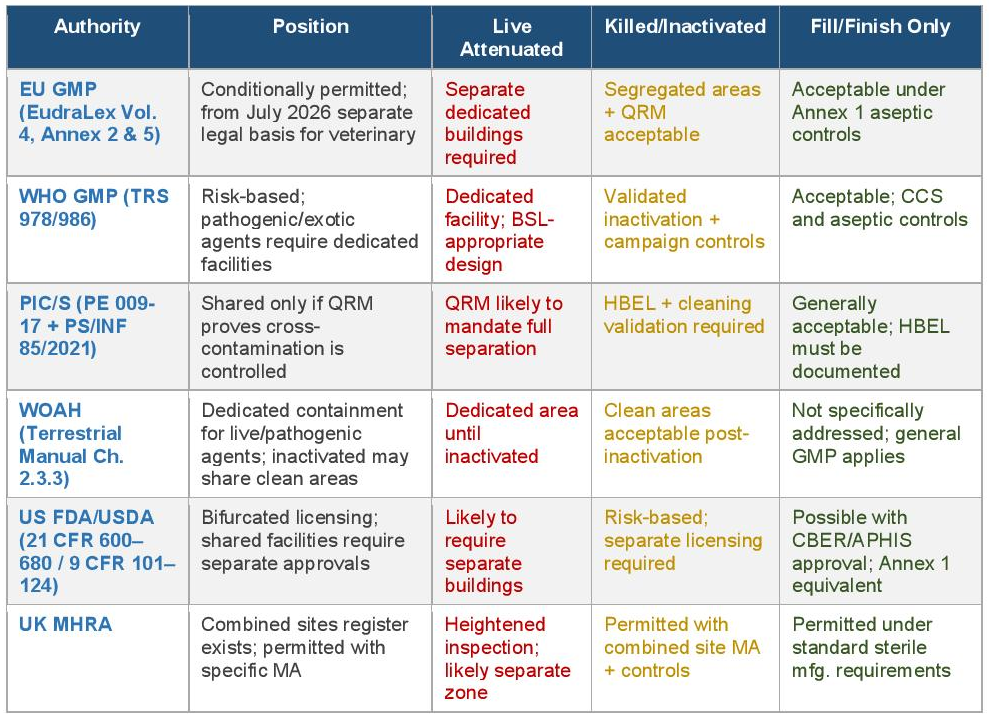
3.1 EU GMP — EudraLex Volume 4 (Annexes 2 and 5)
EU GMP Annex 5 provides the most granular regulatory language directly relevant to the risk dimensions identified above. Several of its provisions map precisely onto the three-dimensional risk framework.
Live agent containment requirement: Live biological agents must be handled in contained areas, with the containment level dependent on the pathogenicity of the micro-organism and whether it has been classified as exotic. [11] This provision explicitly ties containment requirements to pathogenicity — not merely to species of origin — meaning that zoonotic veterinary vaccine strains may require the same or higher containment level as the corresponding human vaccine strain.
Inactivated agent requirement: Inactivated biological agents should be handled in clean areas — not containment areas. [12] This is the regulatory basis for the lower-risk classification of post-inactivation operations and the permissibility of shared clean-area facilities for killed vaccine downstream processing and fill/finish.
Spore-forming bacteria: Production areas where biological agents particularly resistant to disinfection — such as spore-forming bacteria — are handled must be separated and dedicated to that particular purpose until the biological agents have been inactivated. [11] This is an absolute requirement, not subject to QRM override.
Campaign principle: With the exception of blending and subsequent filling operations, one biological agent only should be handled at a time within an area. [11] This principle governs all live-agent upstream operations and applies regardless of whether the agents are human or veterinary, viral or bacterial.
2026 regulatory separation: From 16 July 2026, Commission Implementing Regulation (EU) 2025/2091 establishes a dedicated legal GMP basis for veterinary medicinal products, technically aligned with but legally separate from EudraLex Volume 4. [15] This formalises the dual-framework reality that manufacturers operating combined sites must navigate.
3.2 WHO GMP — Technical Report Series 978 and 986
WHO GMP for biological products (TRS 978, Annex 1) and WHO biosafety risk assessment guidance for vaccine manufacturing (including pathogen-specific guidelines for influenza, polio, and rabies vaccine production) provide a complementary risk-based framework.
The WHO explicitly links facility design requirements to biosafety levels, and its guidelines for specific vaccines illustrate the tiered approach:
- Live attenuated influenza vaccine production: requires BSL-2 enhanced or BSL-3 enhanced facility design, with specific pressure cascade and ventilation requirements. Shared facilities with human/veterinary products are not addressed as acceptable without explicit risk justification.[9]
- Inactivated polio vaccine (IPV): although produced from highly pathogenic poliovirus (BSL-2+ upstream), post-inactivation the product is handled in standard clean-room conditions. The 2017 poliovirus release incident from an IPV manufacturing facility in the Netherlands illustrates that even inactivation-step vaccines require rigorous containment upstream.[10]
- BCG (live attenuated Mycobacterium bovis): WHO guidance and PIC/S GMP both specify dedicated facilities for BCG production. This is among the clearest examples of an absolute dedication requirement for a live attenuated bacterial vaccine.[2]
The WHO’s core principle — that facility requirements must be proportionate to the risk presented by the specific organism, and that those risks must be assessed for both human safety and environmental protection — applies symmetrically to veterinary agents with zoonotic potential. [16]
3.3 PIC/S — PE 009-17 and PS/INF 85/2021
PIC/S guidance on cross-contamination in shared facilities (PS/INF 85/2021) provides the most operationally detailed framework for the QRM assessment that underpins any shared-facility arrangement. It requires manufacturers to document, for each product pair in a shared facility:
- Whether the biological agents involved have zoonotic potential and, if so, what that potential is (species range, transmission routes, pathogenicity in humans);[17]
- Health-Based Exposure Limits (HBELs) or equivalent risk metrics for each biological agent — recognising that HBELs for biological agents are more complex to derive than for chemical entities;[17]
- The adequacy of cleaning and decontamination procedures, including biological activity validation (e.g. viral inactivation, sporicidal efficacy) rather than chemical cleaning validation alone;[13]
- The effectiveness of physical separation, pressure differentials, and HVAC segregation in preventing cross-contamination between production areas.[17]
PIC/S is explicit that where QRM demonstrates that no form of control short of a dedicated, self-contained facility can prevent cross-contamination to an acceptable level, then a dedicated facility is required — regardless of economic considerations. [17] This principle applies with particular force to live attenuated vaccines and to any vaccine involving organisms with significant zoonotic potential.
3.4 WOAH — Terrestrial Manual Chapters 2.3.3 and 1.1.8
WOAH standards — which govern the GMP requirements for veterinary vaccine manufacturing in 182 member countries — provide a direct complement to the EU and WHO frameworks and include several provisions that are directly responsive to the risk dimensions analysed in this article.
WOAH Chapter 2.3.3 explicitly differentiates containment requirements by biological agent status: live agents require contained areas; inactivated agents require clean areas. [12] This maps precisely onto the upstream/downstream risk gradient identified in Section 2.3 above.
WOAH Chapter 1.1.8 requires that methods of production document measures to prevent cross-contamination of lower-risk materials by higher-risk materials. [16] This provision is directly applicable to mixed live/killed manufacturing scenarios and to the co-manufacture of veterinary and human vaccines where one product may present a higher biological hazard than the other.
WOAH also specifically addresses the zoonotic risk dimension: the zoonotic risks of all organisms used in vaccine production must be assessed, regardless of whether the vaccine is intended for human or animal use. [4] This is a pivotal regulatory statement: a veterinary vaccine agent that is also pathogenic to humans imposes human-safety-level containment requirements, not merely veterinary GMP requirements.
3.5 ICH Q9(R1) — Quality Risk Management
ICH Q9(R1) is the methodological backbone of any QRM assessment for shared vaccine manufacturing facilities. It provides the risk assessment tools — FMEA, HACCP, Fault Tree Analysis, Risk Matrices — that regulators expect manufacturers to apply when justifying a shared-facility arrangement. [18]
For the scenarios analyzed in this article, ICH Q9(R1) requires that risk assessment be:
- Proportionate: more rigorous and quantitative for higher-risk scenarios (live attenuated, zoonotic, upstream); simpler and qualitative where risks are demonstrably lower (inactivated, fill/finish only);[18]
- Science-based: drawing on virological, microbiological, and epidemiological data on the specific organisms involved, rather than generic biological product risk classifications;[18]
- Life-cycle: reviewed and updated when products change, when new organisms are introduced, when facility modifications occur, or when surveillance data indicates a shift in risk profile.[18]
The application of ICH Q9(R1) does not automatically permit shared manufacturing — it provides the framework within which the case for (or against) shared manufacturing must be made. For several of the scenarios analyzed below, a rigorous QRM assessment would systematically identify risks that cannot be controlled short of full physical separation.
4. Risk-Stratified Scenario Analysis
The following analysis applies the three-dimensional risk framework and the global GMP requirements reviewed in Section 3 to six representative co-manufacturing scenarios. The scenarios are ordered from highest to lowest biological risk.
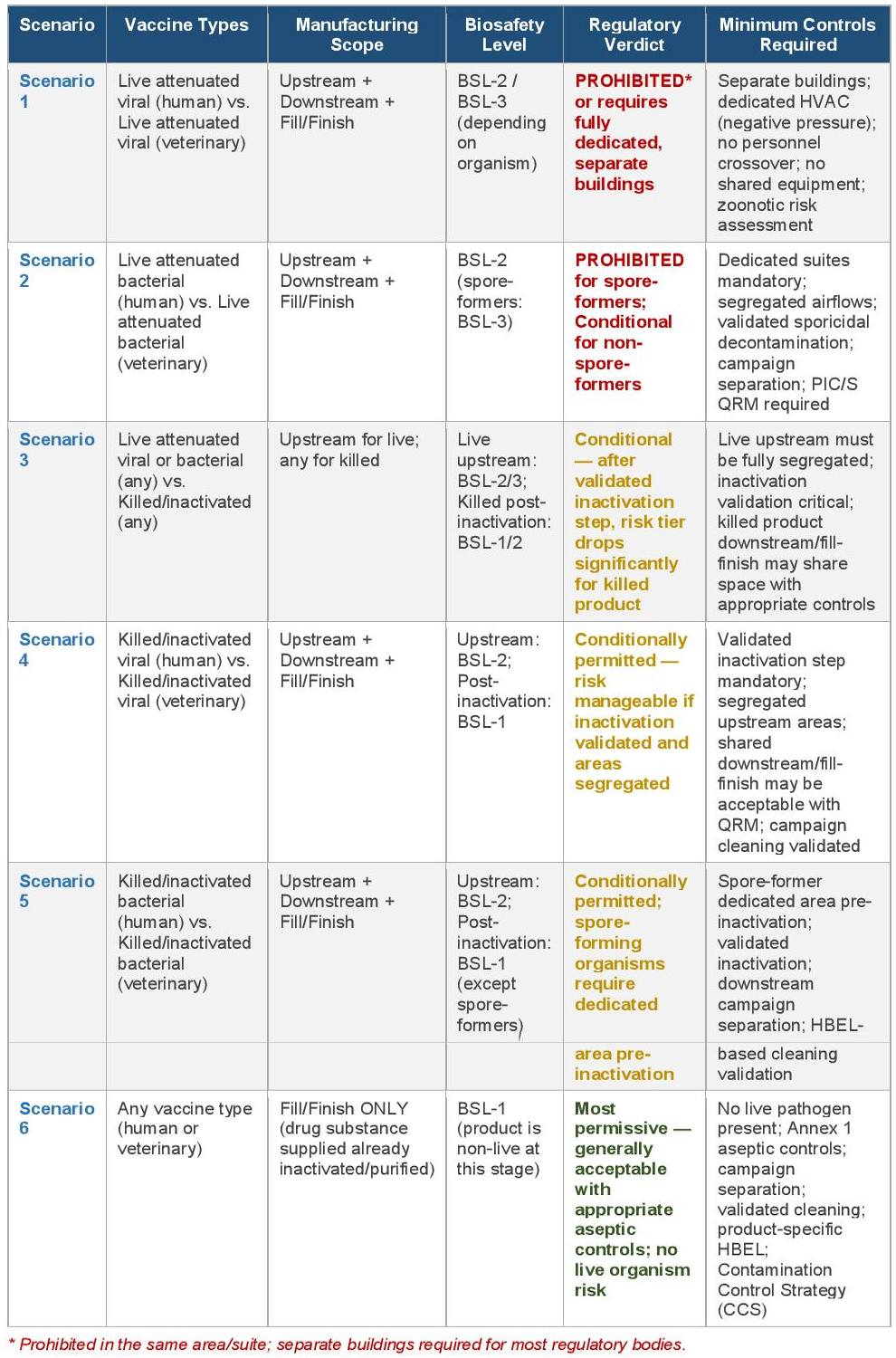
4.1 Scenario 1 — Live Attenuated Viral (Human) + Live Attenuated Viral (Veterinary): Full Manufacturing
RISK LEVEL: HIGH — Generally prohibited or requires fully separate buildings FFE0E0
This is the highest-risk co-manufacturing scenario. Both human and veterinary live attenuated viral vaccine production require BSL-2 (or BSL-3 for highly pathogenic strains) upstream environments. The risks that arise when both are manufactured on the same site are:
- Genetic cross-contamination: live viral strains may recombine if co-present in a facility, potentially generating hybrid organisms with unpredictable pathogenicity or host range;[7]
- Adventitious agent introduction: the veterinary viral strain may carry animal-specific viruses that contaminate the human vaccine process;[7]
- Reversion risk amplified: if both vaccine strains have reversion-to-virulence potential, the combined environmental load increases the probability of reversion events;[3]
- Zoonotic cross-exposure: personnel working with veterinary live attenuated viral strains with zoonotic potential may carry infectious material to human vaccine production areas, even with decontamination procedures.[4]
EU GMP Annex 5 requires that only one biological agent be handled at a time in an area; [11] WHO and WOAH require contained areas for all live agents. [12,16] PIC/S QRM applied to this scenario would systematically identify risks that are not controllable by campaign separation alone, since the two viral strains may circulate in the facility environment between campaigns (in HVAC systems, on surfaces, via personnel vectors) even with robust cleaning programs.
Regulatory conclusion: shared upstream manufacturing of two different live attenuated viral vaccine strains — one human, one veterinary — in the same area or building is incompatible with the GMP frameworks reviewed. Separate dedicated buildings, each with independent HVAC, are the expected standard. The PIC/S QRM assessment burden for a shared arrangement would be extremely high and, in most cases, would conclude that full separation is required.
4.2 Scenario 2 — Live Attenuated Bacterial (Human) + Live Attenuated Bacterial (Veterinary): Full Manufacturing
RISK LEVEL: HIGH — Prohibited for spore-formers; conditionally very restricted for non-spore-formers
Live attenuated bacterial vaccines include some of the most environmentally persistent organisms encountered in pharmaceutical manufacturing. The risk profile differs materially between spore-forming and non-spore-forming species.
Spore-forming bacteria (e.g. Bacillus anthracis in veterinary anthrax vaccines; Clostridium species): EU GMP Annex 5, WOAH Chapter 2.3.3, and WHO guidelines all require that areas handling spore-forming bacteria be separated and dedicated until inactivation is confirmed. [11,12] This is a non-negotiable requirement: validated cleaning of spore-contaminated areas is not achievable to a level that would support re-use for human vaccine manufacture. Shared manufacturing for any product at any stage prior to validated spore inactivation is prohibited.
Non-spore-forming live attenuated bacteria (e.g. BCG [M. bovis], Salmonella Typhi Ty21a, Vibrio cholerae CVD 103-HgR): PIC/S GMP Annex 2 explicitly requires dedicated facilities for BCG manufacture, [2] citing the specific risks of environmental persistence and cross-contamination even at low levels. The quality and safety requirements for live attenuated bacterial vaccines, including the requirement to demonstrate the attenuated phenotype for each batch, impose particular quality control burdens. [2] Co-manufacturing of BCG with any human vaccine — including another bacterial vaccine — is generally considered incompatible with GMP.
4.3 Scenario 3 — Live Attenuated (Any, Upstream) + Killed/Inactivated (Any, Any Stage)
RISK LEVEL: MODERATE-HIGH — Conditional; risk tier drops sharply post-validated inactivation
This hybrid scenario — where one product stream involves live attenuated upstream manufacture and the other involves killed/inactivated agents — is the most nuanced from a risk perspective. The key regulatory question is whether the inactivated product’s manufacturing stages at the shared site are upstream (pre-inactivation), downstream (post-inactivation), or fill/finish only.
If both products have upstream operations at the same site: the live attenuated product’s containment requirements effectively govern the entire shared area during its campaign, and the killed product’s upstream (pre-inactivation) operations are subject to the same live-agent containment requirements as the live vaccine. This scenario requires the same segregation as Scenario 1 or 2 at the upstream level.
If the inactivated product is received at the site as a validated, inactivated drug substance and only downstream or fill/finish operations are performed: the risk profile is substantially lower. The inactivated product presents no live-agent risk, and the key controls are aseptic processing integrity, campaign cleaning, and product mix-up prevention — all manageable within a well-designed multi-product facility. [5,6]
The critical regulatory safeguard in this scenario is the robustness of the inactivation validation for the killed product. Inactivation steps in vaccine manufacturing are classified as critical process steps and must be validated with appropriate safety margins demonstrating complete inactivation of the specific agent. [7,8]
4.4 Scenario 4 — Killed/Inactivated Viral (Human) + Killed/Inactivated Viral (Veterinary): Full Manufacturing
RISK LEVEL: MODERATE — Conditionally permitted with QRM, validated inactivation, and segregated upstream areas
This scenario involves two killed viral vaccine products — one human, one veterinary — with both upstream and downstream operations performed at the same facility. The risk profile is substantially lower than the live attenuated scenarios, but upstream operations still involve live virus at elevated concentrations before the inactivation step.
The upstream phase — cell culture, viral propagation, and harvest — requires BSL-2 conditions for most killed viral vaccines. [3,6] These areas must be segregated between human and veterinary production campaigns, with validated cleaning and decontamination between uses.
The regulatory pivot point is the inactivation step. For killed viral vaccines, inactivation is typically accomplished by treatment with formaldehyde, beta-propiolactone, or physical methods, with validated kinetics demonstrating complete inactivation. [7] Once the inactivation validation package is complete and the process is consistently controlled, downstream operations (purification, formulation) may, in principle, be conducted in shared clean areas under campaign conditions.
The QRM assessment for this scenario must specifically evaluate: (1) the zoonotic potential of the veterinary viral strain — a veterinary virus with significant human pathogenic potential must be treated as if it were a human-pathogenic agent for facility design purposes; [4] (2) the compatibility of the cleaning/decontamination procedures for both agent types; and (3) the HBEL-based assessment of residual antigen carry-over risk between campaigns. [13]
4.5 Scenario 5 — Killed/Inactivated Bacterial (Human) + Killed/Inactivated Bacterial (Veterinary): Full Manufacturing
RISK LEVEL: MODERATE — Conditionally permitted; spore-formers require dedicated area pre-inactivation
Killed bacterial vaccines — including whole-cell pertussis, whole-cell cholera, and various veterinary bacterins — present a distinct risk profile from killed viral vaccines, primarily due to the potential for spore formation and the presence of bacterial endotoxins.
For non-spore-forming killed bacterial vaccines, the risk framework parallels Scenario 4: upstream BSL-2 operations require campaign segregation; post-inactivation downstream operations may potentially share clean areas with appropriate controls.
For killed vaccines derived from spore-forming bacteria: the area must be dedicated until inactivation is confirmed. [11,12] Even though the final product is killed, the upstream production and early downstream phases involve live spore-forming organisms that cannot be removed by standard cleaning. Dedicated areas are mandatory at this stage, precluding concurrent or campaign-shared use with any other vaccine product.
The bacterial endotoxin (pyrogen) cross-contamination risk is specific to this scenario: Gram-negative bacterial vaccines carry high endotoxin loads that can contaminate shared equipment and surfaces in ways that are not addressed by conventional cleaning validation alone. Depyrogenation and endotoxin-specific cleaning validation must be included in the QRM assessment. [13]
4.6 Scenario 6 — Any Vaccine Type: Fill/Finish Operations Only
RISK LEVEL: LOW — Most permissible scenario; generally acceptable under Annex 1 aseptic controls and QRM
Where a facility operates exclusively as a fill/finish site — receiving pre-formulated, validated, inactivated, and purified drug substance from a separate upstream/downstream manufacturing facility — the biological hazard profile is fundamentally different from any of the preceding scenarios.
No live pathogenic organism is present in a fill/finish facility operating on inactivated vaccine drug substance. The contamination risks are those common to any multi-product aseptic manufacturing environment: microbial contamination of the sterile product, mix-up, and residual antigen carry-over between campaigns.
These risks are addressed by the standard GMP controls applicable to aseptic manufacturing under EU GMP Annex 1 (2022 revision): a documented Contamination Control Strategy (CCS); Grade A filling environment within a Grade B background; environmental monitoring; personnel qualification; container closure integrity testing; and product-specific cleaning validation based on HBEL. [14]
The co-filling of human and veterinary vaccine drug substances in the same facility — on a campaign basis — is the scenario most clearly permissible under all reviewed GMP frameworks, provided:
- The drug substances are supplied with documentation confirming validated inactivation/purification;[6]
- Each product has a separate marketing authorisation;[15]
- Campaign separation with validated HBEL-based cleaning is implemented;[13]
- A documented CCS addresses both product types in the multi-product environment;[14]
- Personnel training includes the specific characteristics of both human and veterinary vaccine products.[11]
5. Risk-Stratified Scenario Summary Matrix
The table below provides a consolidated risk-stratified summary of the six co-manufacturing scenarios, mapping vaccine type and manufacturing scope to biosafety level requirements, regulatory verdict, and minimum controls.
6. Regulatory Authority Position by Vaccine Type and Scope
The table below cross-references the position of each major regulatory authority against the three principal risk categories: live attenuated upstream manufacture, killed/inactivated full manufacture, and fill/finish only.
7. Quality Risk Management Implementation for Shared Facilities
7.1 Risk Assessment Methodology
Where a shared-facility arrangement falls into the Moderate or Low risk categories identified above, regulators expect a formal QRM assessment in accordance with ICH Q9(R1). [18] The appropriate methodology varies with the risk level:
- FMEA (Failure Mode and Effects Analysis): most appropriate for systematic evaluation of equipment-level and process-step cross-contamination failure modes in shared downstream or fill/finish operations;[18]
- HACCP (Hazard Analysis and Critical Control Points): appropriate for upstream biological manufacturing, identifying critical control points where biological hazards may be introduced, amplified, or not controlled;[18]
- Fault Tree Analysis: useful for evaluating the combined probability of multiple control failures that would result in cross-contamination or product mix-up in a shared environment;[18]
- Risk Ranking and Filtering: appropriate as a first-pass tool to rank product pairs in a multi-product facility by relative risk, directing more intensive formal assessment to higher-risk combinations.[18]
7.2 Health-Based Exposure Limits for Biological Agents
HBELs are the quantitative foundation for cleaning validation in shared pharmaceutical facilities. For biological vaccine products, HBEL derivation is more complex than for chemical entities: the biological activity of vaccine antigens cannot always be characterised by a simple pharmacological model, and the relevant hazard (immunological response in the wrong species, adjuvant toxicity, endotoxin load) may differ from what HBEL frameworks are designed to address. [13]
Manufacturers must work with toxicologists and clinical/veterinary pharmacologists to derive appropriate acceptance criteria for residual vaccine antigens between campaigns, documenting the derivation in a manner that regulators can review. Where no validated HBEL can be derived — as may be the case for live attenuated organisms — this is itself a finding that supports the requirement for dedicated facilities rather than shared-facility cleaning validation.
7.3 Inactivation Validation as a Risk Control Measure
The inactivation step is a critical process control that, when validated, changes the regulatory risk classification of the manufacturing process and the facility design requirements. Inactivation validation for vaccine manufacturing must demonstrate:
- Complete inactivation of the target organism under defined process conditions (agent concentration, inactivant concentration, temperature, time, pH);[7]
- Robustness across the expected range of process variation, with appropriate safety margins;[7]
- Specificity to the target organism — recognizing that inactivation of the target does not necessarily inactivate all adventitious contaminants.[7][8]
A validated inactivation step does not retroactively justify inadequate upstream containment. Upstream operations involving live agents must be conducted in appropriate containment, regardless of the inactivation step that follows. However, the inactivation validation package is a prerequisite for demonstrating that downstream shared operations present an acceptably low biological risk.
8. Discussion
The risk-stratified analysis presented in this article substantially refines the earlier, undifferentiated regulatory review. Several key conclusions emerge.
First, the vaccine type (live attenuated vs. killed) is the single most important variable. Live attenuated vaccines — viral or bacterial — impose containment requirements that make shared manufacturing with other biological products, whether human or veterinary, extremely difficult to justify under any major GMP framework. Killed/inactivated vaccines, by contrast, transition to a lower risk classification at the point of validated inactivation, opening the regulatory space for shared downstream and fill/finish operations.
Second, the bacterial vs. viral distinction is material primarily in the upstream phase. Spore-forming bacterial vaccines impose an absolute requirement for dedicated areas pre-inactivation that is not present for viral vaccines (which, while requiring containment, are generally more amenable to validated cleaning between campaigns). Post-inactivation, viral and bacterial killed vaccine residues are comparable in their GMP management requirements.
Third, the manufacturing scope is operationally decisive for regulatory permissibility. Fill/finish facilities — operating on inactivated drug substance — are in a categorically different risk position from integrated upstream-downstream facilities. The viability of a shared fill/finish model for human and veterinary vaccines is substantially supported by global GMP frameworks when appropriate campaign controls and aseptic manufacturing standards are applied.
Fourth, zoonotic potential is a cross-cutting risk factor that cuts across all three dimensions. A veterinary vaccine agent with significant zoonotic potential must be treated as a human-pathogenic agent for facility design purposes, regardless of its regulatory classification as a veterinary product. WOAH’s explicit requirement to assess zoonotic risks of all production organisms — whether the vaccine is human or veterinary — is a direct reflection of this principle.
The 2026 separation of EU GMP frameworks for human and veterinary products does not alter the technical risk analysis, but it increases the regulatory compliance burden for combined sites, which will need to demonstrate compliance with two separate legal frameworks simultaneously — each of which cross-references the other for technical alignment.
9. Conclusion
The permissibility of co-manufacturing veterinary and human vaccines in the same facility is not a binary question answerable at the level of ‘vaccine manufacturing’ as a whole. It is a risk-graded question whose answer depends on the intersection of vaccine biological status (live vs. killed), biological agent class (viral vs. bacterial, pathogenicity, zoonotic potential), and manufacturing scope (upstream, downstream, or fill/finish only).
As a practical decision framework:
- Live attenuated vaccines — viral or bacterial — involving upstream manufacture require separate, dedicated facilities or buildings, and shared arrangements with any other vaccine product are not supportable under EU GMP, WHO, PIC/S, or WOAH frameworks;
- Killed/inactivated vaccines with full upstream and downstream manufacture at the same site may share post-inactivation downstream and fill/finish areas with appropriate QRM, campaign controls, and validated cleaning, but must maintain segregated upstream suites;
- Fill/finish facilities operating on pre-inactivated, purified drug substance represent the most permissible co-manufacturing scenario and are generally supportable under all reviewed frameworks with standard aseptic manufacturing controls;
- Spore-forming bacterial organisms impose an absolute dedicated-area requirement pre-inactivation, under EU GMP Annex 5 and WOAH Chapter 2.3.3, that is not subject to QRM override;
- Zoonotic potential of any organism — regardless of whether the vaccine is for human or veterinary use — must be assessed as a specific risk factor, and organisms with significant zoonotic potential must be handled with human-safety-level containment requirements.
Manufacturers planning shared-facility arrangements must engage regulatory authorities proactively and early, grounding their submissions in ICH Q9(R1)-compliant risk assessments that explicitly address these three risk dimensions, the specific organisms involved, and the validated controls applied to each manufacturing stage.
References
[1] Detmer A, Glenting J. Developing and manufacturing attenuated live bacterial vaccines. BioPharm International. 2006;19(9):60–70. Available at: https://www.biopharminternational.com/view/developing-and-manufacturing-attenuated-live-bacterial-vaccines
[2] Pharmaceutical Inspection Co-operation Scheme (PIC/S). PE 009-17: Guide to Good Manufacturing Practice for Medicinal Products, Annex 2 — Biological Medicinal Products for Human Use. Geneva: PIC/S; 2022.
[3] Klessing S, et al. Propagation of SARS-CoV-2 in a closed cell culture device: potential GMP compatible production platform for live-attenuated vaccine candidates under BSL-3 conditions. Viruses. 2023;15(2):397. doi:10.3390/v15020397
[4] Vaccine Insights. Resolving facility design conflicts between biocontainment and good manufacturing practices for vaccines manufacture. Vaccine Insights. 2023;2(4). doi:10.18609/vac/2023/038
[5] NorthX Biologics. Viral vaccine manufacturing for clinical trials. NorthX Biologics Technical Note; 2026. Available at: https://www.nxbio.com/resource/viral-vaccine-manufacturing-for-clinical-trials/
[6] MECART Cleanrooms. Vaccine manufacturing facility design & layout: aseptic fill/finish. Technical Reference; 2025. Available at: https://www.mecart-cleanrooms.com/applications/vaccine-manufacturing-facility-design/
[7] Rele M. Preventing microbial contamination of viral vaccines and vectors. Pharmaceutical Technology. 2025;49(11). Available at: https://www.pharmtech.com/view/preventing-microbial-contamination-viral-vaccines-and-vectors
[8] Nair PM, et al. Strategic and technical considerations in manufacturing viral vector vaccines for BARDA threats. Vaccines. 2025;13(1). PMC11769106. doi:10.3390/vaccines13010075
[9] World Health Organization. WHO Biosafety Risk Assessment and Guidelines for the Production and Quality Control of Human Influenza Pandemic Vaccines. WHO/CDS/EPR/GIP/2007.9. Geneva: WHO; 2007.
[10] Duizer E, et al. Facility-associated release of polioviruses into communities — risks for the posteradication era. PLOS Pathogens. 2019;15(7):e1007922. doi:10.1371/journal.ppat.1007922
[11] European Commission. EudraLex Volume 4, Annex 5: Manufacture of Immunological Veterinary Medicinal Products. Brussels: European Commission; 2004 (as amended). Available at: https://health.ec.europa.eu/document/download/c14c2480-2473-4866-bf2b-d0b7ac61bc27_en
[12] World Organisation for Animal Health (WOAH). Terrestrial Manual, Chapter 2.3.3: Minimum Requirements for the Organisation and Management of a Vaccine Manufacturing Facility. Paris: WOAH; 2024. Available at: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/2.03.03_MANU_SITES_VACCINE_ORG_MANAGE_RQ-CCOIE.pdf
[13] European Medicines Agency (EMA). Guideline on Setting Health-Based Exposure Limits for Use in Risk Identification in the Manufacture of Different Medicinal Products in Shared Facilities. EMA/CHMP/CVMP/SWP/169430/2012. Amsterdam: EMA; 2014.
[14] European Commission. EudraLex Volume 4, Annex 1: Manufacture of Sterile Medicinal Products (2022 revision). Brussels: European Commission; 2022. Specifically: Contamination Control Strategy (CCS) requirements.
[15] European Commission. Commission Implementing Regulation (EU) 2025/2091 of 17 October 2025 laying down good manufacturing practice for veterinary medicinal products in accordance with Regulation (EU) 2019/6. Official Journal of the European Union; 2025 (applicable from 16 July 2026).
[16] World Organisation for Animal Health (WOAH). Terrestrial Manual, Chapter 1.1.8: Principles of Veterinary Vaccine Production. Paris: WOAH; 2024. Available at: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/1.01.08_VACCINE_PRODUCTION.pdf
[17] Pharmaceutical Inspection Co-operation Scheme (PIC/S). PS/INF 85/2021: Cross-Contamination in Shared Facilities — Guidance for GMP Inspectors. Geneva: PIC/S; 2021.
[18] International Council for Harmonisation (ICH). ICH Q9(R1): Quality Risk Management — Step 5 Revision 1. EMA/CHMP/ICH/24235/2006 Corr.2; effective 26 July 2023.
[19] Coronado-Blanco JM, et al. Current GMP standards for the production of vaccines and antibodies: An overview. Frontiers in Public Health. 2022;10:1021905. doi:10.3389/fpubh.2022.1021905
[20] World Health Organization. WHO Technical Report Series No. 978, Annex 1: Good Manufacturing Practices for Biological Products. Geneva: WHO; 2013.
[21] Cleanroom Technology. Vaccine manufacturing facilities and cleanrooms explained: biosafety levels and GMP design. Cleanroom Technology; 2021. Available at: https://cleanroomtechnology.com/vaccine-manufacturing-facilities-and-cleanrooms-explained-176581
